algemeenheid
Cystic fibrosis is de meest voorkomende autosomale recessieve aandoening bij de Kaukasische bevolking en treft ongeveer 1 persoon op elke 2500.
Deze pathologische aandoening staat bekend om zijn schadelijke effecten op de luchtwegen, maar heeft ook invloed op andere systemen zoals het spijsverterings- en voortplantingsstelsel.
Bij personen met cystic fibrosis zijn de luchtwegen verstopt met dik, viskeus slijm, dat moeilijk te verwijderen is, zelfs bij de meest energieke hoest. Ademen wordt moeilijk en patiënten - als er niet voortdurend inspanningen worden gedaan om de luchtwegen meerdere keren per dag schoon te houden - lopen het risico dood te gaan door hun eigen uitscheiding. Cystic fibrosis-patiënten sterven vaak ten gevolge van longontsteking, omdat de verstopte luchtwegen een vruchtbare omgeving bieden voor bacteriegroei.

oorzaken
Cystic fibrosis wordt veroorzaakt door mutaties in het cystische fibrose fibrose (CFTR) transmembraan geleidingsgen dat gelokaliseerd is op chromosoom 7 (locus mapping: 7q31).
Ten minste 1500 mutaties van het CFTR-gen zijn bekend. De meest voorkomende mutatie wordt gewoonlijk "Delta-F508" (DF508) genoemd en wordt bepaald door de deletie van 3 basenparen in exon 10, wat resulteert in het verlies van fenylalanine op positie 508.
Het eiwit gecodeerd door het CFTR-gen is een transmembraankanaal dat behoort tot de ATPase-superfamilie van trafficking of ABC-transporters, gelokaliseerd op het niveau van het apicale membraan van epitheelcellen en verantwoordelijk voor het transport van chloorionen.
Onder normale omstandigheden scheiden bepaalde cellen die langs de luchtwegen lopen, slijm af samen met een waterige vloeistof die hun dichtheid verlaagt. Bij cystische fibrose wordt de afscheiding van de waterige vloeistof sterk verminderd , bijgevolg wordt het slijm zeer dicht en moeilijk te verwijderen uit de luchtwegen.
In het ademhalingsepitheel, net als alle epithelia die vloeistoffen vervoert, hangt het transport van water af van het transport van opgeloste stoffen. Om water uit te scheiden, transporteren de cellen van het respiratoire epitheel actief chloorionen (Cl-) van de interstitiële vloeistof naar het lumen, waardoor een negatieve elektrische potentiaal ontstaat die een passieve natriumstroom (Na +) in dezelfde richting veroorzaakt. De bewegingen van Na + en Cl- verhogen de osmotische druk van de vloeistof die de helling van het epitheel windt naar het lumen, bijgevolg beweegt het water passief volgens de osmotische gradiënt van de interstitiële vloeistof naar het lumen. Het gendefect dat het begin van cystische fibrose beïnvloedt, voorkomt direct het transport van Cl- en interfereert indirect met het transport van Na + en water. Dientengevolge wordt de osmotische gradiënt die nodig is voor de afscheiding van water niet gecreëerd in het epitheel.
Risicofactoren
- Familie erfenis . Gezien het feit dat cystische fibrose een erfelijke ziekte is, die op een autosomaal recessieve manier wordt overgedragen, is het belangrijk om de familiegeschiedenis (anamnese) van toekomstige ouders te beschouwen.
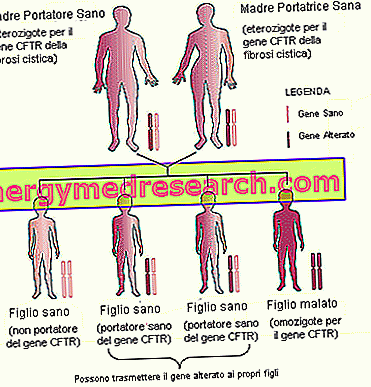
- Ras . De incidentie van cystische fibrose is groter bij blanke mensen in het noorden en van Europese origine.

Symptomen en klinische symptomen
Voor meer informatie: Symptomen van cystische fibrose
De ernst van de symptomen kan variëren, afhankelijk van het verloop van de ziekte: de meeste klinische tekenen beïnvloeden het ademhalingssysteem en het gastro-intestinale systeem.
Ademhalings tekenen en symptomen
Het dikke en viskeuze slijm geassocieerd met cystische fibrose veroorzaakt luchtwegobstructie en remt de activiteit van de trilharen die het longepitheel bekleden, waardoor de eliminatie van het slijm zelf wordt vergemakkelijkt. In de kleine luchtwegen draagt de resulterende slijmerige stagnatie bij aan het creëren van een ideale omgeving voor bacteriën, zoals Streptococcus aureus of Pseudomonas aeruginosa, die het getroffen individu predisponeren voor terugkerende infecties en ontstekingen, die op hun beurt ook ernstig kunnen zijn, zoals longontsteking. De chronische aard van deze ontstekingsprocessen kan longweefsel in de loop van de tijd beschadigen en dientengevolge tot een slechte ademhalingsfunctie leiden.
De kenmerkende symptomen die zich tijdens het verloop van cystic fibrosis ontwikkelen, zijn dus:
- Aanhoudende hoest met sputumproductie
- Hijgend adem
- Kortademigheid en moeite met ademhalen
- Terugkerende longinfecties
Spijsverteringskanaal tekenen en symptomen
Dik slijm kan ook de kanalen afsluiten die spijsverteringsenzymen van de pancreas naar de dunne darm transporteren. Zonder deze spijsverteringsenzymen kan de darm de voedingsstoffen in het ingenomen voedsel niet volledig verteren en volledig opnemen (met name vetten, cholesterol en in vet oplosbare vitaminen A, D, E en K).
Het resultaat is vaak het volgende:
- Gezwollen buik, meteorisme, steatorrhea
- Slechte gewichtstoename en slechte groei (door ondervoeding)
- Intestinale blokkade, vooral bij pasgeborenen (meconium ileum)
- Constipatie of ernstige obstipatie
complicaties
Ademhalingssysteemcomplicaties
- Bronchiëctasie : cystische fibrose is een van de hoofdoorzaken van bronchiectasie, een aandoening die de luchtwegen beschadigt, met als gevolg obstructief ventilatoir falen, met een duidelijke vermindering van de expiratoire flow.
- Chronische infecties : het dikke slijm dat stagneert in de longen en sinussen biedt een ideale voedingsbodem voor bacteriën en schimmels. Dientengevolge kunnen mensen met cystic fibrosis frequente aanvallen van sinusitis, bronchitis of pneumonie hebben.
- Neuspoliepen : aangezien de binnenkant van de neus ontstoken en gezwollen is, kunnen zachte vlezige gezwellen (poliepen) ontstaan die de ademhaling tijdens de slaap kunnen belemmeren.
- Hoest met bloed (bloedspuwing): na verloop van tijd kan mucoviscidose de luchtwegwanden verdunnen. Als gevolg hiervan kunnen patiënten met cystic fibrosis bloed uit de luchtwegen afgeven, meestal door een hoest.
- Pneumothorax : deze aandoening, die bestaat uit de accumulatie van lucht in de pleuraholte, komt vaker voor bij oudere mensen met cystische fibrose. Pneumothorax kan pijn op de borst en kortademigheid veroorzaken.
- Instorting van de longen : chronische pulmonaire infecties kunnen het longparenchym beschadigen, waardoor de kans groter is dat de longen instorten.
- Respiratoire insufficiëntie : na verloop van tijd kan mucoviscidose longweefsel beschadigen en de functionaliteit ervan aantasten.
Complicaties van het spijsverteringsstelsel
- Voedingstekorten : dik slijm kan de alvleesklierkanalen blokkeren die spijsverteringsenzymen transporteren van de pancreas naar de dunne darm. Zonder deze enzymen kan het lichaam geen eiwitten, vetten en in vet oplosbare vitamines opnemen.
- Diabetes : bètacellen van de alvleesklier scheiden insuline af, wat nodig is om de bloedglucoseregulatie te reguleren. Cystic fibrosis verhoogt het risico op het ontwikkelen van diabetes. Ongeveer 20 procent van de getroffen mensen ontwikkelt diabetes op de leeftijd van 30, dus patiënten worden regelmatig getest op bloedglucose.
- Obstructie van de galwegen : het kanaal dat gal van de lever afvoert en van de galblaas naar de dunne darm kan geblokkeerd en ontstoken raken, wat leidt tot leverproblemen en soms galstenen.
- Rectale verzakking : frequente hoest of inspanning tijdens ontlasting (constipatie) kan ervoor zorgen dat het interne rectale weefsel uit de anus steekt.
- Intussusceptie : kinderen met cystic fibrosis lopen een hoger risico op intussusceptie, een aandoening waarbij een deel van de darmplooien zich terugplooien en daardoor invaginaties veroorzaken. Het resultaat is darmobstructie, een noodsituatie die onmiddellijke behandeling vereist.
Complicaties van het voortplantingssysteem
Bijna alle mannen met cystische fibrose zijn steriel, omdat de kanalen die de testikels en de prostaatklier (zaadleider) met elkaar verbinden door slijm of helemaal ontbreken (de afwezigheid van de leidingen is nog steeds een onbekende oorzaak).
Hoewel vrouwen met cystic fibrosis mogelijk minder vruchtbaar zijn dan andere vrouwen, blijven ze toch in staat om zwanger te worden en de zwangerschap tot een einde te brengen. Zwangerschap kan echter bijdragen aan een verslechtering van symptomen van cystische fibrose.
Andere complicaties
Osteoporose : mensen met cystic fibrosis lopen een hoger risico op het ontwikkelen van osteoporose, met als gevolg verlies van massa en botsterkte. Patiënten moeten worden geadviseerd om calcium, vitamine D en bisfosfonaten naar behoefte te gebruiken.
Elektrolyt-onevenwichtigheden : mensen met cystische fibrose hebben meestal een hoger zoutgehalte dan normaal in het zweet. Tekenen en symptomen zoals een toename van de hartslag, vermoeidheid, zwakte en lage bloeddruk helpen om de decompensatie die de hydro-saline balans in het bloed ondergaat te benadrukken.
Diagnose en therapie van cystische fibrose »


