algemeenheid
Craniosynostosis is de term waarmee artsen een abnormaliteit van de schedel aanduiden als gevolg van de voortijdige fusie van één of meer craniale hechtingen.
Craniale hechtingen zijn de vezelachtige gewrichten die de botten van de schedelboog (dwz de frontale, temporale, pariëtale en occipitale botten) samenbinden.
Craniosynostose kan een geïsoleerd fenomeen zijn (niet-syndromale craniosynostose) of het gevolg zijn van bepaalde ziektebeelden (syndromische craniosynostosis). Onder de ziektetoestanden die een voortijdige fusie van de craniale hechtingen veroorzaken, zijn de bekendste: Crouzon-syndroom en Apert-syndroom.
Met de voortijdige fusie van de schedelhechtingen hebben de hersenstructuren niet de geschikte ruimte om te groeien. Dit heeft verschillende consequenties, waaronder voornamelijk de toename van de intracraniale druk (intracraniële hypertensie).
Een snelle en nauwkeurige diagnose maakt het mogelijk om een ad-hocbehandeling te plannen. De laatste is van een chirurgisch type en heeft als laatste doel de scheiding van fusesi hechtingen vroeg.
Herinneringen aan de anatomie van de menselijke schedel
Uitgerust met botten en kraakbeen, is de schedel de skeletstructuur van het hoofd dat het gezicht vormt en de hersenen, het cerebellum, de hersenstam en de sensorische organen beschermt.

Om de studie en het begrip van de schedel te vereenvoudigen, hebben de anatomen gedacht om het in twee compartimenten te verdelen, genaamd neurocranium en splancnocranium .
neurocranium
Het neurocranium is het bovenste craniale gebied, dat de hersenen en enkele van de belangrijkste sensorische organen bevat. De belangrijkste botten - strikt vlak - zijn de voorhoofds-, temporale, pariëtale en occipitale botten; deze, samen genomen, vormen de zogenaamde craniale kluis .
splanchnocranium
Het splanchocranium, of gezichtsmassief, is het antero-inferieure gebied van de schedel, samengesteld uit gelijkmatige en ongelijke botten. Vertegenwoordigt de skeletstructuur van het gezicht, daarom bevat het benige elementen zoals de onderkaak, bovenkaak, jukbeenderen, neusbeen enz.
Wat is craniosynostose?
Craniosynostosis is een zeldzame anomalie van de schedel, gekenmerkt door een onnatuurlijke vorm van het hoofd als gevolg van de voortijdige fusie van één of meer craniale hechtingen . Craniale hechtingen zijn de vezelachtige gewrichten die de botten van de schedelboog (dwz de frontale, temporale, pariëtale en occipitale botten) samenbinden.

Van de site: //www.wkomsi.com/
WANNEER MOET DE SLUITING VAN CRANALE SUTURES WORDEN VERMELD?
Onder normale omstandigheden vindt de fusie van craniale hechtingen plaats in de postnatale periode (NB: sommige processen eindigen zelfs op de leeftijd van 20 jaar). Dit lange proces van fusie zorgt ervoor dat de hersenen kunnen groeien en zich goed kunnen ontwikkelen.
Als, zoals in het geval van craniosynostosis, fusie te vroeg plaatsvindt - dus tijdens prenatale, perinatale * of vroege jeugd - ondergaan de encefale elementen (hersenen, cerebellum en hersenstam) en bepaalde zintuigen (met name ogen) een verandering van vorm en groei.
oorzaken
Het pathofysiologische proces dat craniosynostosis bepaalt, is de voortijdige fusie van schedelhechtingen .
Dit proces kan een geïsoleerd fenomeen vertegenwoordigen - waarbij we met "geïsoleerd" bedoelen dat het niet geassocieerd is met een bepaalde morbide toestand - of het kan het gevolg zijn van bepaalde syndromen, bijna altijd van genetische aard.
In het licht hiervan hebben artsen besloten om craniosynostosis in twee categorieën te classificeren:
- Niet-syndromale craniosynostosis . De term niet-syndromisch geeft aan dat de craniale anomalie niet geassocieerd is met enige pathologie of ander fysiek defect.
- Syndroom craniosynostosis . De term syndroom betekent dat de craniale misvorming het resultaat is van een bepaald syndroom, in de meeste gevallen van een genetisch type.
NIET-SINDROMISCHE CRANIOSINOSTOSE
Artsen en onderzoekers hebben nog niet vastgesteld wat de oorzaken van niet-syndromale craniosynostose zijn.
Ze stelden verschillende hypothesen voor - inclusief de invloed van omgevingsfactoren of problemen van het hormonale type - maar geen van deze theorieën vond bevestiging in de experimentele resultaten.
Om de precieze oorsprong van de anomalie te begrijpen, zijn daarom in dit verband verdere studies nodig.
CRANIOSINOSTOSI SINDROMICA
Volgens het laatste medische onderzoek zijn er meer dan 150 verschillende syndromen, allemaal vrij zeldzaam, die craniosynostosis kunnen veroorzaken.
Van deze syndromen zijn de bekendste en meest voorkomende:
- Crouzon-syndroom . Het resultaat van specifieke mutaties in de genen FGFR2 (chromosoom 10) en FGFR3 (chromosoom 4), deze specifieke morbide aandoening beïnvloedt een pasgeborene om de 60.000 en leidt tot de aanwezigheid van anomalieën exclusief op het niveau van het hoofd en het gezicht.
- Apert-syndroom . Het ontstaat voornamelijk als gevolg van mutaties in het FGFR2-gen (hetzelfde als bij het Crouzon-syndroom) en treft elke 100.000 één pasgeborene.
In tegenstelling tot het Crouzon-syndroom, zijn de genetische veranderingen van FGFR2 zodanig dat misvormingen niet alleen de schedel en het gezicht betreffen, maar ook handen en voeten.
- Pfeiffer-syndroom . Het ontstaat als gevolg van mutaties in het "gebruikelijke" FGFR2-gen en een gen met vergelijkbare functies, genaamd FGFR1 (chromosoom 8). De eigenaardigheid van deze mutaties is dat ze - naast de misvormingen van de schedel en het gezicht - ook bepalen: syndactylie, brachydactylie en duimen en grote tenen (onevenredig aan de andere vingers).
Pfeiffer-syndroom heeft een incidentie van één geval per 100.000 pasgeborenen.
- Saethre-Chotzen-syndroom . Het is een genetische aandoening die elke 50.000 of zo een pasgeborene treft. Veroorzaakt verschillende misvormingen in schedel, gezicht, handen en voeten. Sommige specifieke mutaties van het TWIST1-gen, gelegen op chromosoom 7, zijn verantwoordelijk voor het begin van het Saethre-Chotzen-syndroom.
EPIDEMIOLOGIE VAN CRANIOSINOSTOSI
Op basis van de meest recente statistieken lijkt het erop dat een kind elke 1800-3000 of zo lijdt aan craniosynostosis.
Wat het meest getroffen geslacht betreft, hebben verschillende klinische onderzoeken aangetoond dat 3 van de 4 patiënten mannetjes zijn. De reden waarom craniosynostosis meer voorkomt in de mannelijke populatie is volkomen onbekend.
Craniosynostosis risicofactoren.
- Laag geboortegewicht
- Vroeggeboorte
- Geavanceerde vaderlijke leeftijd
- Moeders roken tijdens de zwangerschap
Symptomen en complicaties
De meeste van de waargenomen symptomen in de aanwezigheid van craniosynostosis zijn te wijten aan een toename van de druk in de schedel . In de geneeskunde wordt de toename van de druk in de schedel intracraniële hypertensie of intracraniële hypertensie genoemd .
In aanwezigheid van craniosynostose is intracraniële hypertensie een gevolg van het feit dat de hersenen en andere structuren in de schedel niet de juiste ruimte hebben om te groeien, dus gaan ze op de benige structuren van het hoofd duwen.
Dit gezegd hebbende, is het belangrijk om te onthouden dat craniosynostosis, als er veel craniale hechtingen bij betrokken zijn of als de aandoening niet op tijd behandeld wordt, kan leiden tot een verminderde ontwikkeling van cognitieve vermogens en een laag IQ.
SYMPTOMEN VAN ENDOCRANISCHE HYPERTENSIE
De mogelijke symptomen van intracraniële hypertensie zijn:
- Aanhoudende hoofdpijn. Over het algemeen wordt het 's morgens en' s nachts erger.
- Visieproblemen. Ze bestaan uit dubbel zien, wazig zicht en wazig zien.
- braken
- prikkelbaarheid
- Gezwollen of prominente ogen
- Moeilijkheden bij het volgen van de beweging van objecten
- Gehoorproblemen
- Ademhalingsproblemen
- Veranderingen in mentale status
- papilledema
Het aantal schedelhechtingen dat betrokken is bij de ontwikkeling van craniosynostose heeft een beslissende invloed op de aanwezigheid van intracraniale hypertensie.
Artsen hebben bijvoorbeeld waargenomen dat de betrokkenheid van een enkele craniale hechtdraad bij 15% van de patiënten intracraniële hypertensie induceert; terwijl de betrokkenheid van ten minste twee hechtingen leidt tot een toename van de druk in de schedel bij ten minste 60% van de patiënten.
In de aanwezigheid van een milde vorm van craniosynostose begint intracraniële hypertensie problematisch te zijn, waardoor de bovengenoemde symptomatologie, ongeveer 4-8 jaar, veroorzaakt wordt.
TEKENS VAN DE CRANIOSINOSTOSI
Onder de tekenen van craniosynostosis zijn de meest voorkomende:- Vormingen van stijve ruggen langs de schedelhechtingen
- Afwijkingen van craniale fontanellen
- Hoofd met afmetingen die niet in verhouding staan tot de rest van het lichaam
SOORTEN CRANIOSINOSTOSE
De vorm van het hoofd van patiënten met craniosynostosis hangt af van welke craniale hechtingen voortijdig zijn gesloten.
Na dit te hebben opgemerkt, achtten artsen het passend om craniosynostosis in verschillende typen te onderscheiden, afhankelijk van de betrokken craniale hechtingen.
De soorten craniosynostosis zijn:
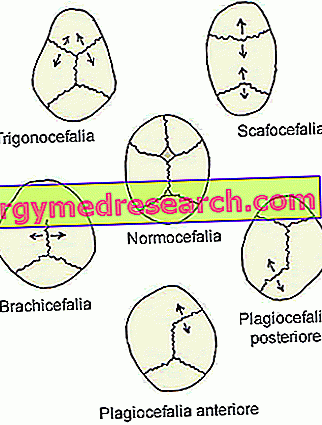
- Sagittaal synostosis ( dolichocephaly of scaphocephaly ). Het is het meest voorkomende type craniosynostose; in feite kenmerkt het ongeveer de helft van de klinische gevallen.
Zijn aanwezigheid valt samen met de voortijdige sluiting van de sagittale craniale hechtingen, gelegen in het bovenste deel van de schedel, tussen de pariëtale botten.
Van //en.wikipedia.org/wiki/Plagiocephaly
- Coronale craniosynostose ( brachycefalie ) Het is het tweede meest voorkomende type craniosynostose; kenmerkt ongeveer elke klinische casus.
Het begin omvat de voortijdige fusie van de coronale hechtingen, die lopen tussen het voorhoofdsbeen en de pariëtale botten.
- Metopic synostosis ( trigonocephaly ). Het is een zeer zeldzame vorm van craniosynostosis, die slechts 4-10% van de gevallen onderscheidt.
Het uiterlijk valt samen met de voortijdige versmelting van de metopische (of frontale) hechtdraad, die van de neus naar het bovenste deel van het hoofd loopt, waarbij het voorste bot in tweeën wordt gescheiden. Over het algemeen wordt deze hechtdraad natuurlijk ingebed in het zesde levensjaar.
- De lambdoïde synostosis ( plagiocephaly ). Het is het zeldzaamste type craniosynostosis. In feite is het slechts 2-4% van de klinische gevallen.
De aanwezigheid ervan omvat de vroege fusie van de lambdoïde hechting, die zich bevindt tussen de pariëtale botten en het achterhoofdsknobbel, achter in het hoofd.

COMPLICATIES
Naast een afbreuk aan de intellectuele ontwikkeling, kan een onbehandelde craniosynostose bepalen:
- Het zogenaamde obstructieve slaapapnoesyndroom .
- Permanente gelaatsveranderingen, vooral in de ogen en oren.
- Permanente misvormingen aan de basis van de schedel (bijvoorbeeld de misvorming, of Arnold-Chiari-syndroom).
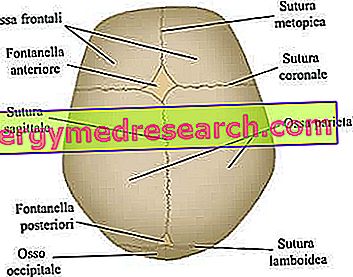
De belangrijkste craniale hechtingen die betrokken zijn bij het craniosynostoseproces. Van de site: www.sciencebasedmedicine.org
- Hydrocephalus .

diagnose
Om craniosynostose te diagnosticeren, zijn lichamelijk onderzoek, evaluatie van de klinische geschiedenis en radiologische beelden van röntgenfoto's of CT-scans naar het hoofd van essentieel belang.
Als de craniosynostose van het syndromische type is, is het ook belangrijk om de morbide aandoening vast te stellen die tot het begin leidde. Daarom konden artsen bloedtesten doen en vooral ook genetische counseling .
ONDERZOEKDOELSTELLING
Het lichamelijk onderzoek bestaat uit een zorgvuldige analyse door de arts van de klinische symptomen op het hoofd van het subject, waarvan wordt vermoed dat ze aan craniosynostose lijden.
Over het algemeen is een kinderarts verantwoordelijk voor het uitvoeren van deze belangrijke diagnostische controle.
KLINISCHE GESCHIEDENIS
De evaluatie van de klinische geschiedenis is belangrijk voor diagnostische doeleinden, omdat het vragen bevat die verband houden met de risicofactoren van craniosynostose.
Daarom zal de arts (meestal altijd een kinderarts) onderzoeken of:
- De baby wordt te vroeg geboren of van een laag gewicht.
- Wat was de leeftijd van de vader op het moment van de conceptie.
- Als de moeder rookt tijdens de zwangerschap.
RADIOLOGISCHE TESTS
Röntgenfoto's en CT in het hoofd dienen meer dan wat dan ook om de diagnose te bevestigen en de arts te laten zien welke schedelhechtingen voortijdig zijn gefuseerd.
De kennis van welke craniale hechtingen erbij betrokken zijn, maakt het plannen van de meest geschikte chirurgische behandeling mogelijk.
behandeling
Craniosynostosis is alleen te genezen door chirurgische ingreep .
Dit laatste bestaat uit een operatie van scheiding van de schedelhechtingen die vroegtijdig uit elkaar versmelten.
Het uiteindelijke therapeutische doel van chirurgie is om de encefale structuren en sommige zintuigen, zoals de ogen, te voorzien van die ruimte die nodig is om zich op zijn best te ontwikkelen en te functioneren.
DE BESTE TIJD VOOR INTERVENTIE
Er bestaat geen volledige overeenstemming, door artsen, over de beste tijd om een craniosynostose-operatie uit te voeren.
Volgens sommige experts zou de ideale periode voor de operatie zijn in de late kindertijd, wanneer het risico op een recidief lager is (dwz een tweede voortijdige fusie van de schedelhechtingen). In geval van een terugval moet de operatie in feite worden herhaald en dit wordt niet aanbevolen gezien de fijnheid van de procedure.
Volgens andere experts zou de meest geschikte tijd zijn in de vroege kinderjaren (tussen 6 en 12 maanden van het leven), wanneer de schedel nog niet volledig is verbeend en de botten nog steeds kneedbaar zijn. De mogelijkheid om botten te vormen (kneedbaarheid) maakt het mogelijk om eventuele morfologische afwijkingen van de botten op te lossen, wat ernstige esthetische defecten en functionele problemen (aan de kaak of ogen) op een meer volwassen leeftijd zou kunnen veroorzaken.
MOGELIJKE CHIRURGISCHE BENADERINGEN
Er zijn twee verschillende chirurgische benaderingen: traditionele chirurgie, ook wel "open" genoemd, en endoscopische chirurgie.
- Traditionele (of "open") operatie .
Het voorziet in algemene anesthesie (daarom is de patiënt tijdens de hele operatie buiten bewustzijn) en de praktijk van een chirurgische incisie op het hoofd, precies op het punt waar de radiologische beelden de schedelanomalie vertoonden.
Door de incisie op het hoofd verwijdert de opererende chirurg (een neurochirurg) het abnormale bot en vertrouwt het toe aan een specialist in craniofaciale chirurgie, die het modificeert en het een vorm geeft die de normale ontwikkeling van de hersenstructuren mogelijk maakt.
Na de modificatie plaatst de neurochirurg het bot opnieuw in de oorspronkelijke positie en sluit de incisie met steken.
Zoals veel traditionele chirurgie, is de "open" operatie enigszins invasief; om het echter voordelig te maken, is het feit dat het in staat is om de botstructuur nauwkeurig en met goede resultaten te modificeren.
- Endoscopische chirurgie interventie .
Het gaat om het gebruik van een endoscoop, een hulpmiddel vergelijkbaar met een flexibele buis, uitgerust met een glasvezelcamera, aan een uiteinde en aangesloten op een monitor.
Vanuit operationeel oogpunt bestaat het uit het inbrengen van de endoscoop in een opening gemaakt op de schedel en in de scheiding, door middel van de endoscoop zelf, van de gesmolten hechtdraad vroegtijdig.
De neurochirurg weet zich in het hoofd te oriënteren, dankzij de beelden die de camera projecteert op de extern aangesloten monitor.
De endoscopische chirurgische interventie is beslist minder invasief dan de "open" operatie (de ziekenhuisperiode is ook korter), maar het heeft twee nadelen: het is alleen aangegeven voor patiënten van enkele maanden (6 in het algemeen), de die kneedbare botten bezitten; loopt een groter risico op herhaling.
POSTOPERATIEVE FASE
Over het algemeen moet een patiënt met craniosynostose, die een operatie moet ondergaan, ongeveer 4-5 dagen na de operatie in het ziekenhuis blijven. Gedurende deze tijd monitoren de neurochirurg en zijn stafleden periodiek hun vitale functies en controleren ze of alles goed gaat.
Na het aftreden is een reeks periodieke controles gepland, die eerst halfjaarlijks zijn en vervolgens, met de groei van de patiënt, jaarlijks.
prognose
De prognose is afhankelijk van verschillende factoren, waaronder:
- De oorzaken die craniosynostosis veroorzaakten. Sommige genetische ziektes die verantwoordelijk zijn voor deze anomalie zijn zeer ernstig en hebben een slechte prognose.
- De positie van de hechtingen versmelt vroeg. Als de hechtingen zich bevinden op posities die voor de neurochirurg "oncomfortabel" zijn om te bereiken, wordt de interventie van craniosynostose gecompliceerd en levert deze mogelijk niet de gewenste resultaten op.


