algemeenheid
Amyloïdose is de term die wordt gebruikt om een groep ziekten te definiëren die wordt gekenmerkt door de accumulatie, vaak in het extracellulaire gebied, van een fibrillair proteïnemateriaal, gedefinieerd als amyloïde . Onoplosbare amyloïde fibrillen vormen bijzonder stabiele afzettingen in veel organen.

Andere foto's Amyloidosis - Galerij 2
De symptomen en de ernst van de ziekte zijn afhankelijk van het orgaan dat voornamelijk wordt beïnvloed door de accumulatie van amyloïde en het type amyloïdose. De meeste gevallen zijn echter systemisch. Met andere woorden, fibrillaire afzettingen zijn wijdverspreid en kunnen mogelijk de functie van veel weefsels en organen van het lichaam in gevaar brengen. De diagnose wordt bepaald door een biopsie, waarbij een klein weefselmonster onder een microscoop wordt onderzocht. Potentiële etiologische factoren variëren afhankelijk van de variant van amyloïdose. De beschikbare behandelingen helpen om de symptomen te beheersen en de productie van amyloïde te beperken.
Kenmerken van amyloïde afzettingen
Amyloïdose is het gevolg van aandoeningen van de secundaire structuur van eiwitten (met een β-gevouwen leaflet-configuratie). Onder normale omstandigheden worden de eiwitten in feite gesynthetiseerd in een lineaire reeks van aminozuren, die door vouwen een specifieke ruimtelijke conformatie aanneemt (eiwitvouwing). Dankzij de structuur, dus de juiste eiwitvouwing, is het eiwit in staat om de fysiologische functies uit te voeren waarvoor het wordt gebruikt. De? Amyloïde proteïnen zijn afgeleid van een precursor die door de cellen verkeerd is verwerkt (vanwege het "verkeerd vouwen"). De fibrilvormende eiwitten zijn gediversifieerd volgens grootte, aminozuursequentie en natieve structuur, maar ze worden onoplosbare aggregaten die qua structuur en eigenschappen vergelijkbaar zijn. De fibrilvoorlopers worden weergegeven door primaire moleculen (bijvoorbeeld: immunoglobuline lichte keten, ß2-microglobuline, apolipoproteïne Al enz.) Of uit producten die een verandering in de aminozuursequentie weerspiegelen. De afwijkende secundaire structuur maakt een predispositie voor de vorming van fibrillen, die plaatselijk in weefsels en organen kunnen worden afgezet en die tot verslechtering van hun normale fysiologische functie kunnen leiden. Er zijn meer dan 20 verschillende eiwitprecursoren geïdentificeerd die een amyloïdconformatie kunnen aannemen, daarom zijn er veel verschillende soorten amyloïdose.
Op basis van de locatie van amyloïde afzettingen kan de ziekte worden onderscheiden in:
- Gelokaliseerde vorm: beperkt tot een bepaald orgaan of weefsel (hart, nieren, gastro-intestinaal stelsel, zenuwstelsel en dermis) en is gewoonlijk minder ernstig dan systemische (diffuse) vormen. Amyloïdose kan bijvoorbeeld alleen de huid aantasten, waardoor depigmentatie en / of jeuk optreedt. Een bepaald type amyloïde-eiwit is ook gevonden in de hersenen van patiënten met de ziekte van Alzheimer. Gelokaliseerde amyloïdose is typisch voor senescentie en van patiënten met type 2-diabetes (waarbij eiwit zich ophoopt in de pancreas).
- Systemische vorm: amyloïde afzettingen zijn aanwezig in verschillende organen en herkennen in het algemeen de neoplastische, inflammatoire, genetische of iatrogene oorsprong. Systemische amyloïdose is vaak erg ernstig: het beschadigt het hart, de nieren, de darmen en de zenuwen en veroorzaakt progressief orgaanfalen.
classificatie
Er zijn veel vormen van amyloïdose, ingedeeld naar de aard van de eiwitten die fibrillaire afzettingen vormen.
De meest voorkomende varianten zijn:
- Primaire amyloïdose (ook lichte amyloïdose genoemd, AL);
- Secundaire amyloïdose (ook wel verworven amyloïdose, AA);
- Erfelijke amyloïdose;
- Amyloïdose geassocieerd met veroudering (of seniele systemische amyloïdose).
AL-amyloïdose
Zie ook: Amyloïdose symptomen
De meest voorkomende vorm van systemische amyloïdose is de primaire (AL). AL-amyloïdose wordt veroorzaakt door de accumulatie van fibrillen die lichte ketens van immunoglobulinen bevatten die zijn afgeleid van monoklonale plasmacellen. De ziekte is vaak een gevolg van monoklonale gammopathieën en kan worden geassocieerd met multipel myeloom of andere lymfoproliferatieve aandoeningen van de B-keten.
Bij AL-amyloïdose kunnen fibrillaire afzettingen zich geleidelijk aan vestigen op het niveau van de organen vele jaren vóór de klinische presentatie. De symptomen zijn variabel en hangen naast de algemene verschijnselen (vermoeidheid, oedeem en gewichtsverlies) af van het orgaan dat het meest is aangetast en van de grootte van de amyloïde afzettingen. Als deze bijvoorbeeld in de nieren worden afgezet, kunnen ze chronisch nierfalen veroorzaken, terwijl als ze in het hart zijn gelokaliseerd, ze het vermogen om voldoende bloed aan het hele lichaam toe te dienen, kunnen schaden. Het amyloïde is typisch gelokaliseerd in de nier, het hart, de lever, het perifere zenuwstelsel en het autonome zenuwstelsel. Andere districten die mogelijk worden getroffen zijn: longen, huid, tong, schildklier, darm en bloedvaten.
AL-amyloïdose kan mogelijk de volgende symptomen veroorzaken:
- Zwakte en aanzienlijk gewichtsverlies;
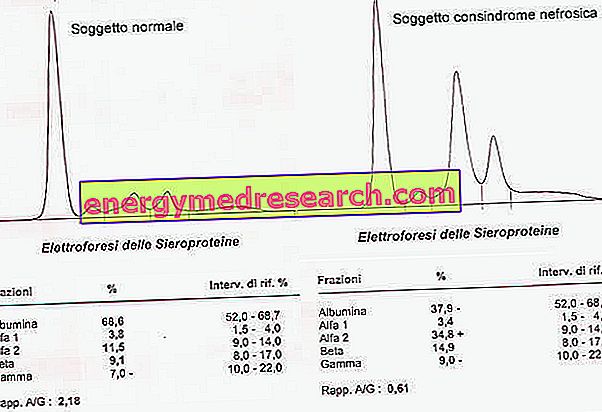
- Vochtretentie met oedeem (als gevolg van hartfalen of nefrotisch syndroom);
- duizeligheid;
- Kortademigheid;
- Gevoelloosheid of een tintelend gevoel in de handen en voeten;
- Carpaal tunnel syndroom (verstoring van de mediane zenuwfunctie);
- Huidletsels: petechiën en ecchymosen;
- Paars rond de ogen;
- Macroglossia (volumetrische groei van de tong.).
Secundaire, seniele en erfelijke amyloïdose
De minder vaak voorkomende vormen van systemische amyloïdose worden hieronder kort beschreven:
- Secundaire amyloïdose (AA) : het is ook bekend als verworven amyloïdose en kan zich ontwikkelen als een complicatie van verschillende ziekten die een aanhoudende inflammatoire aandoening veroorzaken (zoals tuberculose, reumatoïde artritis, lepra en familiale mediterrane koorts) en bepaalde vormen van kanker (bijvoorbeeld: celcarcinoom nier). Bij deze processen stimuleert de interventie van pro-inflammatoire cytokines (IL-1, IL-6 en TNF) de hepatische productie van serumamyloïde A (SAA). SAA kan worden gevonden in hoge concentraties in het serum van patiënten met reumatoïde artritis, de ziekte van Crohn en in erfelijke vormen van periodieke koorts, ten minste tot de ontstekingsfase van deze chronische aandoeningen is verzwakt. Typische plaatsen voor accumulatie van amyloïde zijn milt, lever, nieren, bijnieren en lymfeklieren. Secundaire amyloïdose presenteert in feite op kenmerkende wijze proteïnurie en / of hepatosplenomegalie, om de betrokkenheid van deze organen aan te tonen. Het behandelen van de onderliggende aandoening voorkomt vaak dat amyloïdose verergert.
- Seniele systemische amyloïdose (cardiaal) : amyloïdose geassocieerd met het normale verouderingsproces wordt over het algemeen gevonden bij patiënten ouder dan 60 jaar. De afzettingen, in deze vorm, worden gedeponeerd op cardiaal niveau. De oorzaken zijn nog niet bekend en nieuwe diagnostische tests en behandelingen zijn momenteel in ontwikkeling.
- Erfelijke amyloïdose : is in sommige families waargenomen als het gevolg van een genetisch defect. Deze mutaties zijn van invloed op specifieke bloedeiwitten (zoals het transthyretine-eiwit, TTR) en kunnen op autosomaal dominante wijze worden overgeërfd. Erfelijke amyloïdose betreft voornamelijk het zenuwstelsel: patiënten ontwikkelen een kenmerkende symmetrische sensorimotorische neuropathie in hun onderste ledematen. Andere amyloïde afzettingen bevinden zich op het niveau van het hart, de bloedvaten en de nieren.
Mogelijke effecten van fibrillaire afzettingen bij amyloïdose
| Aangetast orgaan of systeem | Mogelijke gevolgen |
| hersenen | De ziekte van Alzheimer |
| Spijsverteringsstelsel | Macroglossie, slikmoeilijkheden, diarree of obstipatie, darmobstructie en slechte opname van voedingsstoffen |
| hart | Hartritmestoornissen (hartritmestoornissen) en hartfalen |
| niertjes | Ophoping van vocht in de weefsels en oedeem, eiwitten in de urine (gedetecteerd door de urinetest) en nierfalen |
| lever | Hepatomegalie (vergrote lever) |
| longen | Ademhalingsproblemen |
| lymfeklieren | Gezwollen lymfeklieren |
| Zenuwstelsel | Carpaal tunnel syndroom, gevoelloosheid, tintelingen of een gebrek aan gevoel in de voeten en op hun plant of een branderig gevoel in deze regio's |
| huid | Papules, purpura en ecchymoses |
| schildklier | Schildklieruitbreiding (schildklierstruma) |
Wie loopt het meeste risico?
Mensen met het volgende profiel hebben een verhoogd risico op het ontwikkelen van amyloïdose:
- Mannelijk geslacht: amyloïdose treft voornamelijk mannelijke proefpersonen;
- Patiënten ouder dan 60;
- Aandoeningen die plasmacellen beïnvloeden (multipel myeloom, lymfoom, monoklonale gammopathie of Waldenström's macroglobulinemie);
- Chronische infectie- of ontstekingsziekte (zoals reumatoïde artritis, inflammatoire darmziekte, familiale mediterrane koorts of spondylitis ankylopoetica);
- Langdurige dialyse;
- Genetische mutaties die de conformatie van eiwitten beïnvloeden.
diagnose
De opeenhoping van grote hoeveelheden amyloïde kan de normale werking van veel organen veranderen. De diagnose van amyloïdose kan zeer uitdagend zijn, omdat de symptomen vaak generiek zijn. Artsen kunnen echter amyloïdose vermoed wanneer:
- Verschillende orgels vertonen een functioneel tekort;
- Vochtretentie vindt plaats, met als gevolg oedeem, op het niveau van de weefsels;
- Een onverklaarbare bloeding treedt op, vooral in de huid (ecchymose, purpura, enz.).
Om andere aandoeningen uit te sluiten, kan de arts beginnen met:
- Lichamelijk onderzoek (om de klinische tekenen van orgaanbetrokkenheid te detecteren);
- Bloed- en urinetests (om het betrokken fibrillaire eiwit te vinden).
De diagnose kan definitief worden bevestigd door biopsie en microscopisch onderzoek van het genomen en verwerkte monster met Congo-rode kleur. Bij sommige patiënten, waarbij amyloïdose wordt vermoed, kan een biopsie worden uitgevoerd op het peri-labische vetkussen. Als alternatief kunnen artsen dezelfde procedure uitvoeren door een nier-, rectaal of huidmonster te nemen. Na de diagnose kan de arts verdere periodieke onderzoeken plannen om de niveaus van verwante stoffen, de grootte en locatie van amyloïde afzettingen, het beloop van de ziekte en de effecten van de behandeling te controleren.
Prognose en therapie
De therapie moet worden aangepast aan de verschillende vormen van amyloïdose, rekening houdend met de basisvoorwaarden en die secundair aan de pathologie. Helaas zijn therapeutische protocollen voor het verminderen of beheersen van de symptomen en complicaties van de ziekte slechts bescheiden succesvol voor de meeste mensen. Geen behandeling gericht op amyloïde afzettingen is nog steeds beschikbaar en de therapie is daarom bestemd voor de onderdrukking van de onderliggende plasmaceldyscrasie, met ondersteunende maatregelen om de functie van het orgaan te ondersteunen en mogelijk te behouden.
Primaire amyloïdose, al dan niet geassocieerd met multipel myeloom, vertoont een slechte prognose en overleving is ongeveer 2-4 jaar. De meeste mensen die aan beide ziekten lijden, overlijden binnen 1-2 jaar. De meest voorkomende doodsoorzaken worden vertegenwoordigd door afbeeldingen van hartfalen, nier- en ademhalingsinsufficiëntie, bloeding uit het maagdarmkanaal en infecties. In het geval van primaire amyloïdose is het hoofddoel de pathologische kloon te erven. Voor dit doel kan chemotherapie worden overwogen. Net als bij de klassieke vormen van monoklonale gammopathieën, worden melfalan of cyclofosfamide (chemotherapeutische middelen die ook worden gebruikt om sommige neoplasma's te behandelen) en dexamethason, een corticosteroïd gebruikt voor zijn ontstekingsremmende effecten, toegediend. De combinatie van deze geneesmiddelen remt abnormale beenmergcellen, zodat het kan leiden tot een geleidelijke vermindering van de hoeveelheid amyloïde in het lichaam en schade aan organen kan voorkomen. Onderzoekers bestuderen andere chemotherapiebehandelingen die geschikt zijn voor de behandeling van amyloïdose. Verschillende geneesmiddelen die worden gebruikt bij de behandeling van multipel myeloom (bortezomib, thalidomide en lenalidomide) zijn ook getest om hun werkzaamheid in de behandeling van amyloïdose te evalueren.
Stamceltransplantatie kan in sommige gevallen een therapeutische optie zijn. Geselecteerde patiënten kunnen effectief worden behandeld met hooggedoseerde intraveneuze toediening van melfalan, gevolgd door transfusie van perifere stamcellen, dwz onrijpe bloedcellen die eerder zijn verzameld ter vervanging van het aangetaste of beschadigde beenmerg (hematopoietisch weefsel).
In het geval van secundaire amyloïdose (AA), vertraagt of verlaagt de behandeling van basale ontstekingsziekte (ontstekingsaandoeningen, chronische infecties of carcinoom) het verloop van de ziekte. De prognose van secundaire amyloïdose hangt af van hoe de onderliggende aandoening wordt behandeld en de overleving is ongeveer 5-10 jaar.
Orgaantransplantatie (nier, hart, enz.) Kan de overleving van een beperkt aantal patiënten met orgaanfalen secundair aan amyloïdose verlengen. De ziekte blijft echter vorderen en zelfs het getransplanteerde orgaan kan amyloïde afzettingen accumuleren (het kan, indien mogelijk, worden vermeden door chemotherapie van het beenmerg te onderdrukken). De uitzondering wordt vertegenwoordigd door levertransplantatie, die de progressie van erfelijke amyloïdose kan beperken (vaak wordt het eiwit dat deze vorm van amyloïdose veroorzaakt gesynthetiseerd in de lever). Het perspectief voor de laatste vorm varieert afhankelijk van het type genmutatie en de mate van voortgang op het moment van diagnose. Amyloïde afzettingen in een specifiek deel van het lichaam kunnen soms operatief worden verwijderd.